Longitudinal follow-up of HPV16 sequence after cervical infection: Low intrahost variation and no correlation with clinical evolution
Abstract
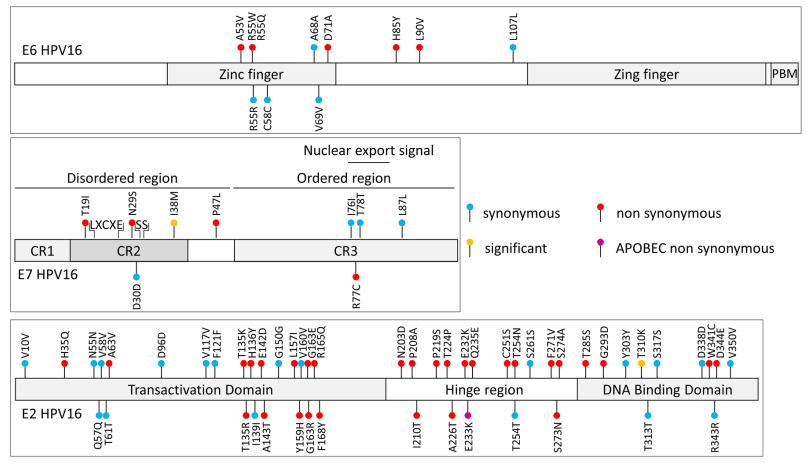
Human papillomavirus (HPV) 16 exhibits different variants that may differ in their carcinogenic risk. To identify some high-risk variants, we sequenced and compared HPV16 whole genomes obtained from a longitudinal cohort of 34 HPV16-infected women who had either spontaneously cleared their infection (clearance group or “C”), or developed cervical high-grade lesions following a viral persistence (group persistence or “P”). Phylogenetic analysis of paired samples obtained at the beginning (C0 or P0) and at the end (C2 or P2) of the follow-up (median intervals between C0–C2 and between P0–P2 were 16 and 36.5 months, respectively) revealed a low genetic variability within the host compared to the genetic interhost diversity. By comparing our HPV16 sequences to a reference sequence, we observed 301 different substitutions, more often transitions (60.9%) than transversions (39.1%), that occurred throughout the viral genome, but with a low frequency in E6 and E7 oncogenes (10 and 9 substitutions), suggesting a high conservation of these genes. Deletions and insertions were mostly observed in intergenic regions of the virus. The only significant substitution found between the subgroups C2 and P2 was observed in the L2 gene (L330F), with an unclear biological relevance. Our results suggest a low longitudinal intrahost evolution of HPV16 sequences and no correlation between genetic variations and clinical evolution.